重点課題2:電子状態・動力学・熱揺らぎの融和と分子理論の新展開
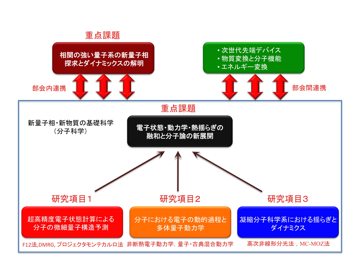 分子理論では、従来、量子化学、特に、電子状態理論、動力学、さらに統計力学や古典論に基づく分子シミュレーションにより、化学事象が各々の側面から明らかされてきた。しかし、分子や分子集団による化学事象の本質的解明のためには、これらが錬成・融合された分子理論が不可欠である。本課題研究では、電子状態・動力学・熱揺らぎの取り扱いをコアエレメントとして個々の革新的な発展を図り、それらを基礎とした融和的な理解と練成による新しい分子理論の展開を行う。特に、数年内で二桁以上ものCPUコアの増加が予想される超並列計算環境は分子科学分野にパラダイムシフトをもたらし、それに最適な物質計算手法の確立は即効性のある多大な成果をもたらすと考えられる。大振幅揺らぎを伴う光合成のエキシトン移動やタンパク質中の無輻射過程といった、これまでの分子理論では困難であった電子と核とが複雑に絡み合った協奏的な量子状態とダイナミクスの取り扱いにより、分子の未知の描像と新しい機能を解き明かし、その理論先導的制御を達成する。その目標のために、本重点課題では以下の三つの研究項目が有機的に連携し、分子論の新機軸を展開する。
分子理論では、従来、量子化学、特に、電子状態理論、動力学、さらに統計力学や古典論に基づく分子シミュレーションにより、化学事象が各々の側面から明らかされてきた。しかし、分子や分子集団による化学事象の本質的解明のためには、これらが錬成・融合された分子理論が不可欠である。本課題研究では、電子状態・動力学・熱揺らぎの取り扱いをコアエレメントとして個々の革新的な発展を図り、それらを基礎とした融和的な理解と練成による新しい分子理論の展開を行う。特に、数年内で二桁以上ものCPUコアの増加が予想される超並列計算環境は分子科学分野にパラダイムシフトをもたらし、それに最適な物質計算手法の確立は即効性のある多大な成果をもたらすと考えられる。大振幅揺らぎを伴う光合成のエキシトン移動やタンパク質中の無輻射過程といった、これまでの分子理論では困難であった電子と核とが複雑に絡み合った協奏的な量子状態とダイナミクスの取り扱いにより、分子の未知の描像と新しい機能を解き明かし、その理論先導的制御を達成する。その目標のために、本重点課題では以下の三つの研究項目が有機的に連携し、分子論の新機軸を展開する。
- 研究項目1:超高精度電子状態計算による分子の微細量子構造予測
- 研究項目2:分子における電子の動的過程と多体量子動力学
- 研究項目3:凝縮分子科学系における揺らぎとダイナミクス
これらの三研究項目のうち、1は次世代電子状態理論の開発を担い、2は多体量子動力学を、3は揺らぎとダイナミクスを担当する。電子状態のF12法、密度行列繰り込み群法、プロジェクターモンテカルロ法、動力学の非断熱動力学、量子−古典混合動力学、熱揺らぎの高次非線形分光法、多中心分子性 Ornstein-Zernike (MC-MOZ)法を出発点に、それぞれの計算手法を連携・発展させ新しい分子理論を推進する。これらの関連課題の関係を下図に示す。目的が異なるが手法として深い関連を持つ部会内(新量子相・新物質の基礎科学)の他の重点課題とも緊密な連絡を図る。更に、本重点課題及びコアを成すサブ重点課題で得られた結果を源流として、次世代先端デバイス・物質変換と分子機能・エネルギー変換との部会間連携を図り次世代スーパーコンピュータを用いた物質計算科学の諸問題の成果をもたらす奔流へと力強い発展を行う。
この重点課題のもとに以降の研究項目での研究を推進する。
研究項目1:超高精度電子状態計算による分子の微細量子構造予測
[担当者] 神戸大:天能精一郎、 分子研:柳井毅、大塚勇起、江原正博、名古屋大:安田耕二、首都大:波田雅彦
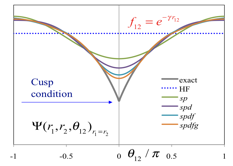
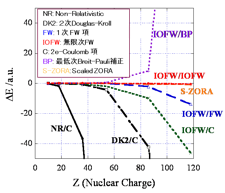
[課題内容・背景・重要性] 計算機が発明されてから約半世紀もの間、分子軌道法による電子状態計算は飛躍的な発展を遂げ、材料設計や創薬といった様々な分野で貢献して来た。一方、これまでの分子軌道法は大規模な二電子反発積分とそのテンソル積のために急激に加速する超並列計算環境で威力を発揮できないという問題や、擬縮重系の取り扱いの困難さがある。数十万コアという超並列計算環境で分子の安定性や励起状態を高精度で計算可能な計算技術の確立は、これまでのアーキテクチャーでは不可能であった様々な物質変換や機能、エネルギー問題、生命現象の解明を実現するものであり、次世代の計算物質科学の本質的な発展に不可欠である。 既存の分子軌道法に取って代わる、超並列環境で実行可能な分子系の大規模・高精度電子状態計算法を確立し、世界をリードする基盤技術の革新的な飛躍と発展を目指す。応用課題との連携を図り次世代スーパーコンピュータを用いた化学反応制御・分子発光・エネルギー問題等の成果へとボトムアップ的に発展させる。 超並列計算環境に適した求積法ベースの電子状態計算手法を用い、ペタフロップ級の実行性能を達成する。高精度F12法による高次電子相関と相対論的手法を駆使し、実験と真に競合する分光学的精度を達成する。更に、高精度の光異性化反応や磁性の研究に必要な相関の強い擬縮重電子状態についても、密度行列繰り込み群(DMRG)法やプロジェクターモンテカルロ法を用いて、超並列計算に適した手法を確立する。
 [計算手法] F12法、密度行列繰り込み群、プロジェクターモンテカルロ法
[計算手法] F12法、密度行列繰り込み群、プロジェクターモンテカルロ法
[次世代スパコンの必要性・実現可能性] F12法はJST-CRESTにより開発が進められて来たGellan量子化学プログラムで並列実装が行われており、理研RICCではMPIのみで20原子程度の二次摂動計算が1000コア以上で並列効率を落とす事無く実行されている。GellanプログラムのF12実装では求積点の数にスケールし、次世代スパコンではハイブリッド並列を用いて百原子程度のナノ分子の完全基底関数極限が数十万コアCPUで計算可能である。DMRG法についても並列実装が既に行われており、プロジェクターモンテカルロ法も超並列向けの手法であるので、強相関電子系についても次世代スパコンを高度に利用した研究の実現可能性が期待出来る。
[具体的な成果目標]次世代スーパーコンピュータで初めて可能になる大規模・高精度計算を実行する。F12理論に基づく二次摂動法で10万コア以上の並列計算を実現化し、金属内包フラーレンなどのナノ分子の完全基底関数極限とその構造最適化による電子状態・構造の解明を行う。更に、F12理論に高次結合クラスター法と相対論を組み合わせる事により、全エネルギーで数10μハートレーの精度を達成する。これにより、分子の超微細量子構造を正確に予測し、実験研究と真に競合する計算物質科学を実現する。更に、DMRGとプロジェクターモンテカルロ法を超並列環境で実行する事により、百電子/百軌道のスケールで強く相関している物質の計算を可能にし、多核金属錯体や光受容体の電子状態を精密に解明する。
研究項目2:分子における電子の動的過程と多体量子動力学
[担当者] 東京大:高塚和夫、東北大:河野裕彦
[課題内容・背景・重要性] 化学動力学は、フェムト秒(10-15秒)の時間スケールでおきる超高速化学反応を直接追跡すべく、原子核の波動関数の時間変化を直接検出する技術や理論を発展させてきた。本課題では、現代化学の基本的概念を提供してきたボルン‐オッペンハイマー描像(質量および時間スケールの大きな相違故、電子と原子核の運動を分離して考える分子像)を超え、また、フェムト秒スケールの原子核波束反応動力学をさらに一歩進め、原子核の運動と強くカップルする電子波束動力学の展開を中心に据え、次世代のアト秒(10-18秒)超高速化学反応電子動力学と反応制御の理論を開拓することを目的とする。現在の多体分子科学は、大きな分子、たとえばタンパクの動的構造転移などを古典動力学シミュレーションによって追究することが盛んに行われている。一方、平衡系においては、モンテカルロ経路積分法などにより静的な量子効果をかなり大きな系にまで考慮することができるようになっている。しかしながら、大きな分子の反応ダイナミクス(原子核の運動のダイナミクス)については、実時間量子動力学の実現は全くと言ってよいほど未開拓な状態におかれている。もちろん、これは、波動関数の短波長性の取り扱いの困難さからきている。本課題では、多体量子動力学および多体量子効果の方法論と計算技術を飛躍的に発展させる。
[計算手法] 量子化学計算に基づいて電子配置関数を発生し、それを基底関数として原子核とのキネマッティックな相互作用を入れて、線形に電子波束を時間発展させる。また、多体量子論としては、これも独自に開発された半古典力学に従って、多体量子波動関数の古典極限を発生させる。
[次世代スパコンの必要性・実現可能性] 電子動力学の研究では、アト秒スケールで電子波束を時間発展させていくが、化学現象が原子核の位置の変位として現れるにはフェムト秒オーダーの長さが必要なため、超高速の計算環境が必要である。また、電子波束波は、通常、電子配置関数で展開されるために、分子の大きさに伴って、大規模計算が必要となる。ただ、この種の計算では、原子核の運動経路に沿った電子混合動力学の形式をとることが多く、経路積分と同じ意味で並列計算が可能である。(ただし、一つ一つの経路で電子状態混合を行う)。多体場に浸された量子系の動力学においても、計算の構造は、非断熱電子動力学と同じである。 また、多体量子動力学では、経路積分の部分的古典化を行うことになるので、やはり並列計算による高速化のメリットが非常に大きい。 本課題研究の骨格をなす非断熱電子・原子核波束動力学の理論と多体半古典力学は、アルゴリズム化され、MPIで結合された500CPU程度のクラスター環境を使って実証研究がなされている。今後、OpenMPによるスレッド並列とMPIによるプロセス並列を併用したハイブリッド並列を利用したアルゴリズム開発を進める。
[具体的な成果目標]
a) 電子・核同時動力学と強レーザーによる電子状態制御
本研究では、現代物理化学の鍵を握っていると考えられる幾つかの具体的な実験事実や理論的問題意識をとりあげ、定性的および定量的に解析・予測をしていく。しかし、個別の成果とは別に、期待される最大の成果は、ポテンシャルエネルギー曲面の概念が破綻する諸現象に対して、新しい理論的方法論を提供し、次世代の研究分野を開拓していくことである。理論化学は、非断熱電子動力学に対する理論的枠組みをようやく手にし、新しい局面に入ろうとしている。Born-Oppenheimer近似を超えた新しい理論により、複数の電子状態が絡む系のダイナミクスにおける電子と核のエンタングルメント・デエンタングルメントの量子相を明らかにし、分子の知られざる姿と性質を引き出す。また、分子に強いレーザーを分子に照射すると、電子と原子核の間に働く引力と同程度の力を外から作用させることができる。このような極限状況では、ボルン‐オッペンハイマー近似は容易に破れ、ベクトルポテンシャル中の電子‐核同時動力学を研究する必要がある。逆にこれを利用して、強い場を使った電子状態の制御を通した化学反応学を創造する。
b) 多体場に浸された量子系の動力学
例えば溶媒中におかれた化学反応系の量子動力学を想定する。ここで環境をなす多体場を古典動力学で近似すると、この系はいわゆる量子・古典混合表示における動力学をなす。これまで量子・古典混合動力学は直観的な取扱いがなされていたが、最近、完全な一般理論が構築された。それは、上に述べた非断熱電子動力学の一部の枠組みと数学的に同等である。この理論を、溶媒中での化学反応動力学などの普遍的な応用研究へと発展させる。
c) 化学動力学における超多次元多体量子動力学理論の開発
化学反応を支える環境場、すなわち溶媒や表面・界面、タンパクが作る局所場、包摂化合物の反応場、クラスター内化学反応などの量子動力学的効果は重要である。この様な系に適用可能な超多次元半古典力学の開発が実用レベルで進んでいる。この方法論では、多次元経路積分よりは圧倒的に少ない計算量で量子効果を計算できるが、なお大規模な並列計算環境を必要とする。これを実現して、古典動力学から量子動力学へと研究のステージを質的に転換させる。
研究項目3:凝縮分子科学系における揺らぎとダイナミクス
[担当者] 分子研:斉藤真司、京都大:佐藤啓文、名古屋大:笹井理生
[課題内容・背景・重要性]溶液や生体系を始めとする凝縮分子系では、分子運動の複雑な絡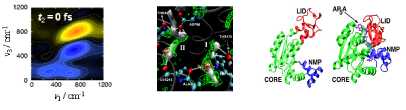 み合いにより、様々な時間・空間スケールをもつ構造揺らぎ・変化または化学反応が誘起され、多様な物性や機能の発現へとつながる。本課題では、複雑液体や生体分子における分子運動の揺らぎ・構造変化の解析を進め、さらに揺らぎや構造変化と反応・機能の関わりの解明を目的とする。物質や生物における様々な機能や性質は、分子の電子状態変化から分子集合体・集団の構造変化に至るまで多層の階層構造を成すダイナミクスが複雑に絡み合った結果として発現する。このような階層性は不均一系を始めとするより複雑な物質系一般に見られる普遍的な構造であり、高度な分光法・一分子分光法を始めとする様々な実験により解析が進められているが、時間・空間スケールの大きく異なる階層構造を貫く我々の理解は非常に限られている。そこで本課題では、溶液やイオン液体さらには蛋白質等の不均一系における揺らぎ・構造変化や化学反応の遅いダイナミクスの理解を目指す。物質や生物における構造形成・機能発現を生み出す分子論的起源の解明は凝縮分子系における重要課題であり、本課題は広く化学過程一般を解析する理論的根幹を供するものと期待される。
み合いにより、様々な時間・空間スケールをもつ構造揺らぎ・変化または化学反応が誘起され、多様な物性や機能の発現へとつながる。本課題では、複雑液体や生体分子における分子運動の揺らぎ・構造変化の解析を進め、さらに揺らぎや構造変化と反応・機能の関わりの解明を目的とする。物質や生物における様々な機能や性質は、分子の電子状態変化から分子集合体・集団の構造変化に至るまで多層の階層構造を成すダイナミクスが複雑に絡み合った結果として発現する。このような階層性は不均一系を始めとするより複雑な物質系一般に見られる普遍的な構造であり、高度な分光法・一分子分光法を始めとする様々な実験により解析が進められているが、時間・空間スケールの大きく異なる階層構造を貫く我々の理解は非常に限られている。そこで本課題では、溶液やイオン液体さらには蛋白質等の不均一系における揺らぎ・構造変化や化学反応の遅いダイナミクスの理解を目指す。物質や生物における構造形成・機能発現を生み出す分子論的起源の解明は凝縮分子系における重要課題であり、本課題は広く化学過程一般を解析する理論的根幹を供するものと期待される。
[計算手法]平衡および非平衡シミュレーションを利用した高次非線形分光法の計算や多時間相関関数の観点から液体等における運動の階層性の理解を深化させる。また、MC-MOZ方程式と分子動力学法を空間的・動的に接合する新規手法により、通常の分子シミュレーションでは解析が困難な蛋白質等の非常に遅い構造変化や自由エネルギーを明らかにし、不均一な系における化学反応ダイナミクスと機能発現の解明を進める。さらに、蛋白質の複数低エネルギー構造を無撞着に参照することにより、大規模な構造転移を記述するポテンシャル関数を導入し、蛋白質のミリ秒以上の遅い構造変化による化学反応の制御を解析する。
[次世代スパコンの必要性・実現可能性]高次非線形分光法に関係する多時間相関関数および無撞着モデルを用いた蛋白質構造転移の解析おいては数千万から数億の構造配置での構造サンプリングが必要であり、MC-MOZ方程式においては多中心分割において並列計算が必要となるため、次世代スパコンやそれに準ずる計算資源の利用が求められる。これらの基礎理論はアルゴリズム化され小規模の並列環境で実証研究がなされており、今後アルゴリズム開発を進める。
[具体的な成果目標]溶液や生体系など凝縮分子系に潜む複雑な運動を多時間相関関数の計算により解析を行い、緩和ダイナミクスや相関した不均一ダイナミクス等を解明する。また、自由度による運動の時間スケールの分離を考慮した新規手法および無撞着モデルにより、通常の分子シミュレーションでは解析が困難な非常に遅い反応における構造変化や自由エネルギー、蛋白質による化学反応制御の機構を明らかにする。このように、凝縮分子系における揺らぎ・構造転移を有効に解析する理論を開発し、揺らぎや構造変化と反応や機能との関わりに対する新しい理解とその設計指針を創出する。